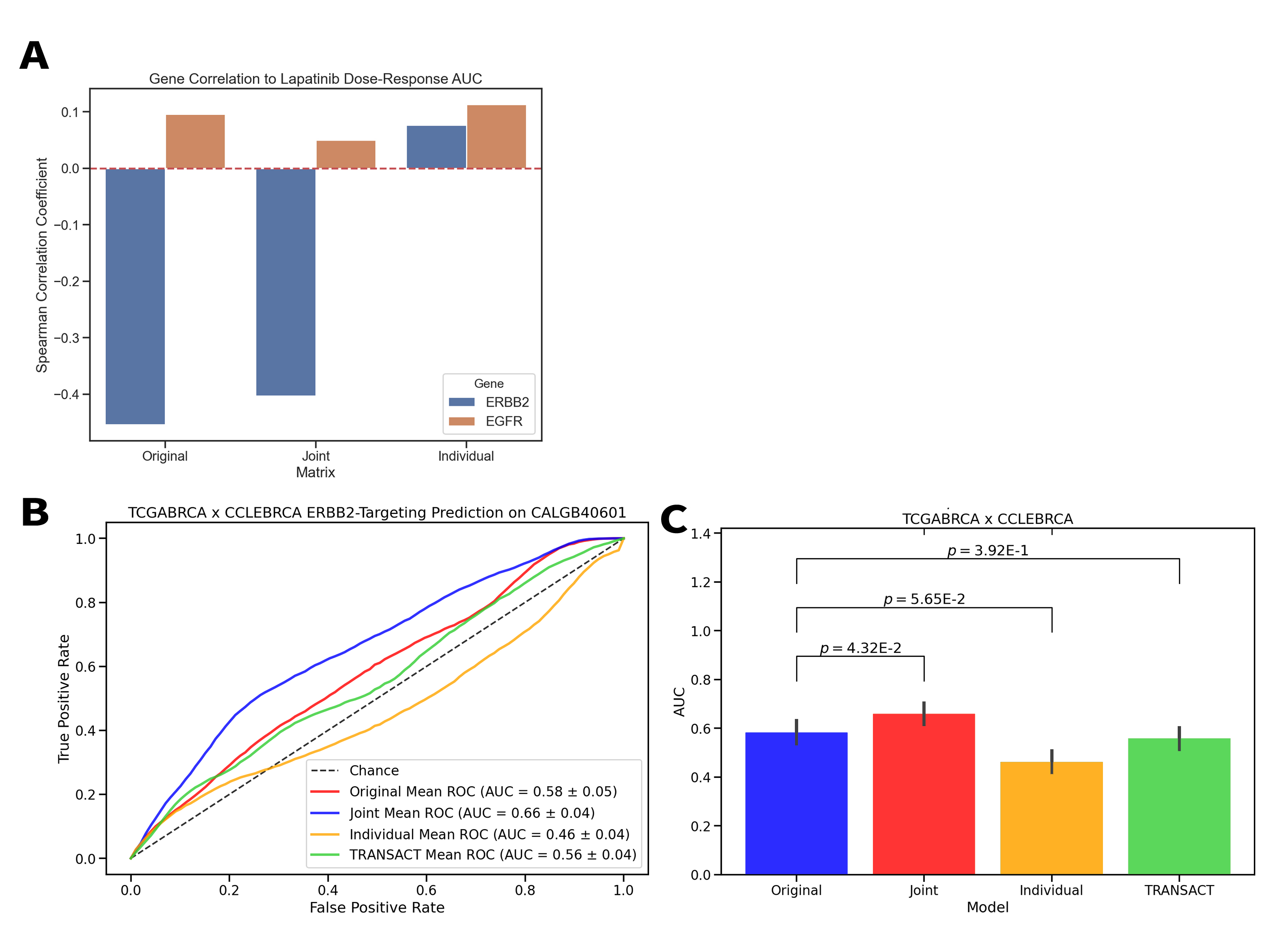
Translating Model System Genomics to Clinical Populations through Joint Dimension Reduction
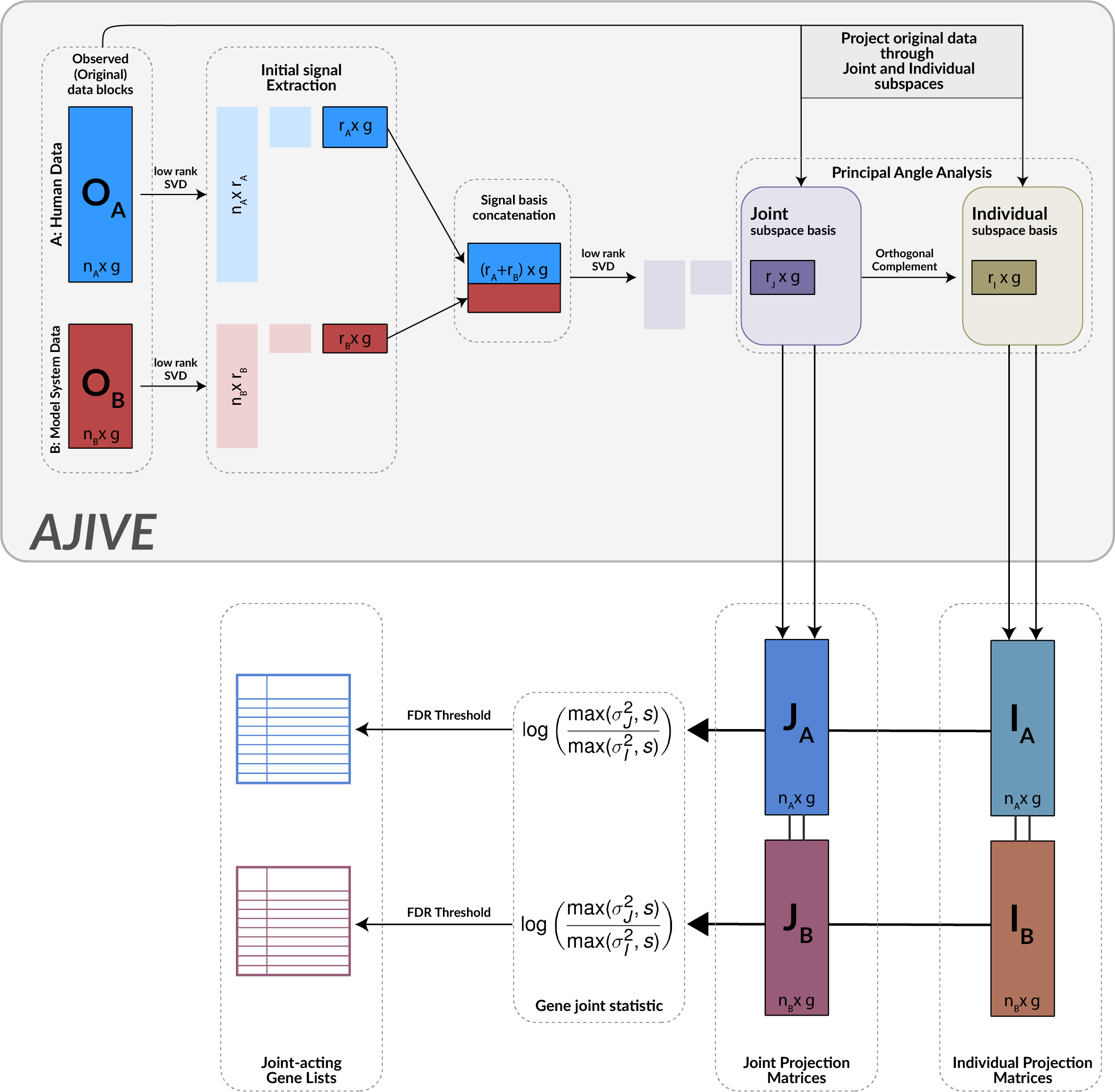
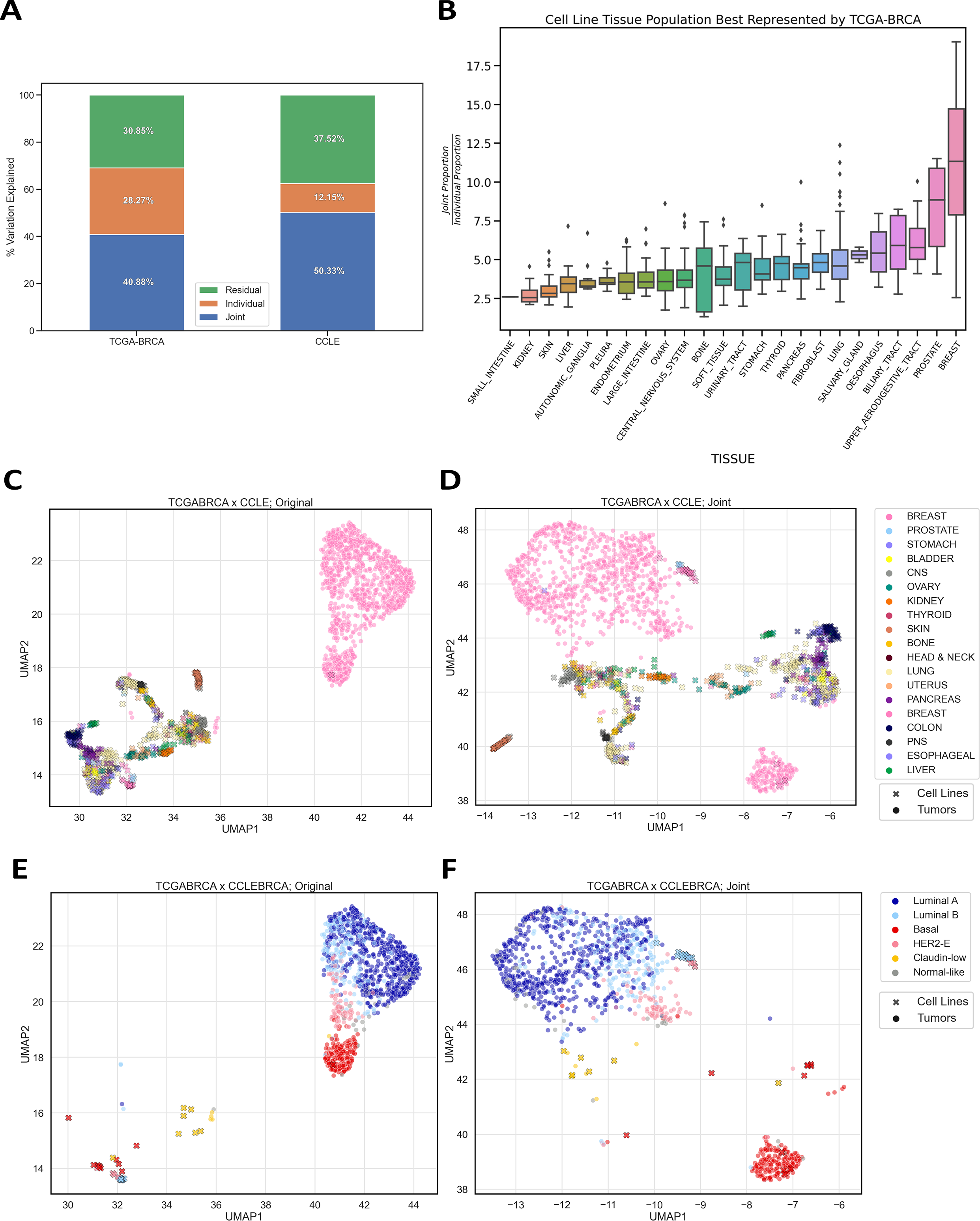
Model systems are an essential resource in genomics research. They simulate effects that we can infer into humans, but come at a risk of inaccurately representing human biology. This inaccuracy can lead to inconclusive experiments or misleading results, urging the need for an improved process for translating model system findings into human-relevant data. I am investigating applications of joint dimension reduction (jDR) to horizontally integrate gene expression data across model systems and human tumor cohorts. By identifying joint-acting modes of variation, we can significantly improve how predictive models and biomarkers are translated into clinical populations.
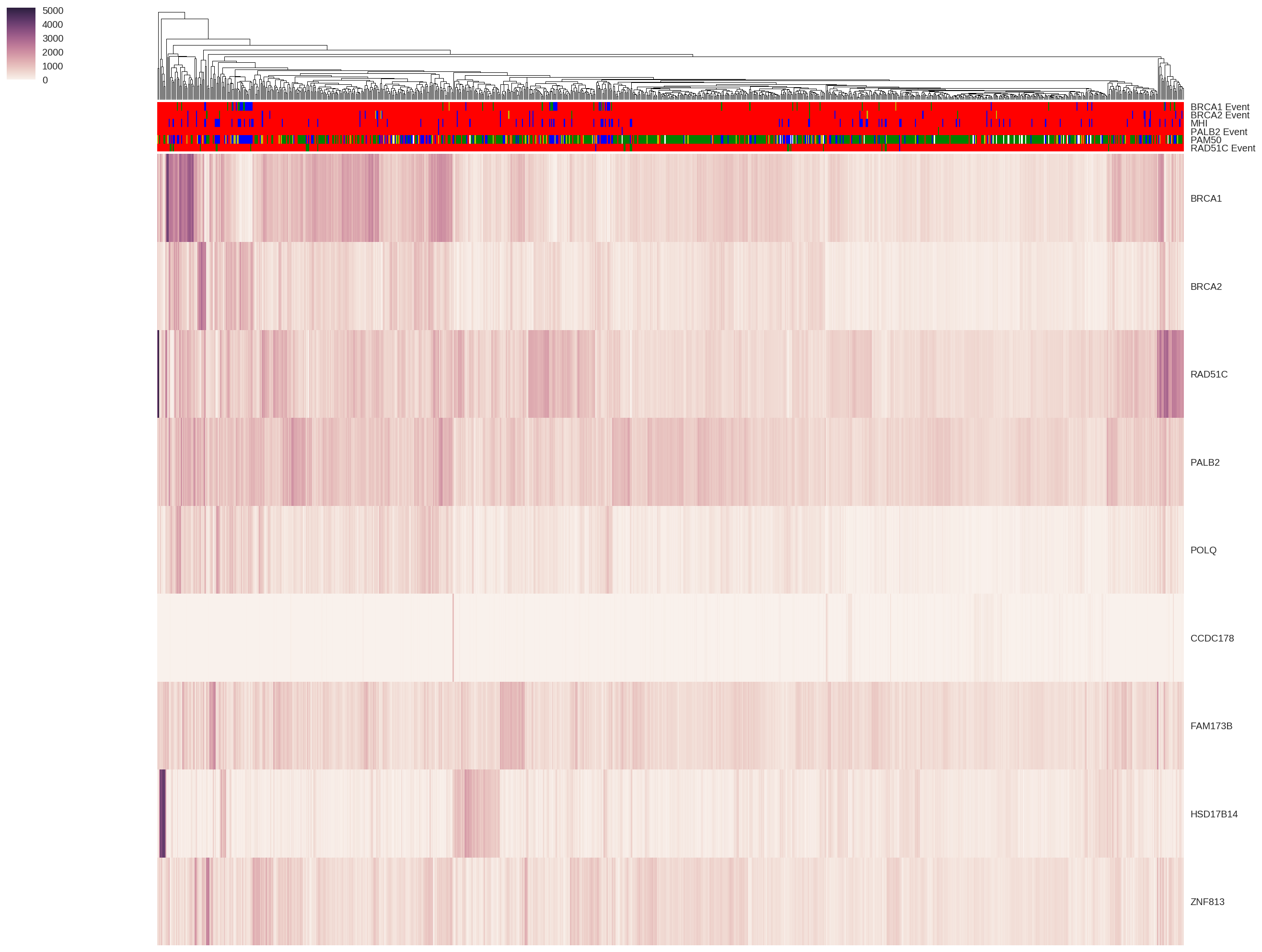
Predicting Predispositions to Error-prone DNA Repair Pathways
Tumors located in the same physical region can have vastly different attributes and respond to treatment in very different ways. I am investigating how our germline DNA can set the foundation for how these different tumors could develop, and how we can better guide treatment for a patient's specific tumor. My work involves understanding how preferences towards certain error-prone DNA repair pathways could result from inherited genetic variation.
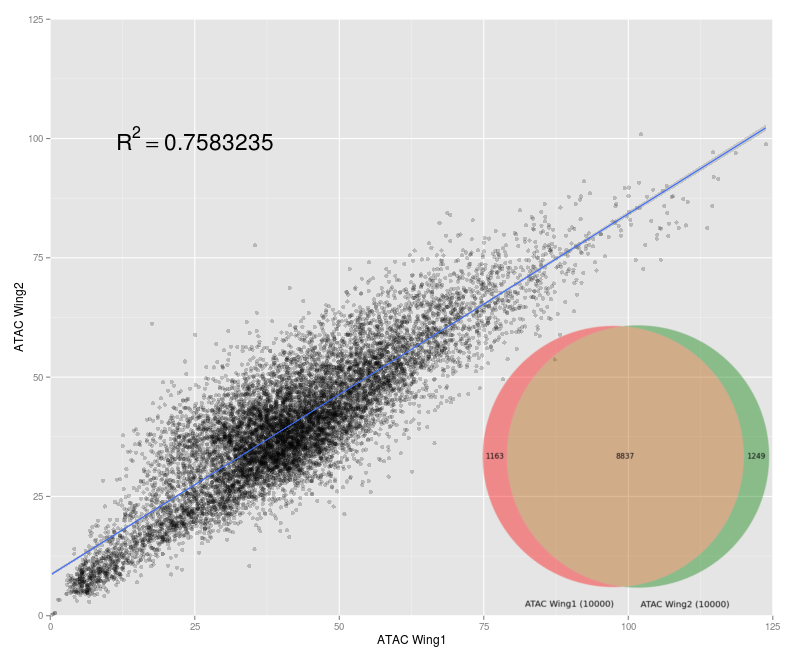
ATAC-seq Data Analysis and Quality Control
ATAC-seq is the most current method for probing chromatin accesibility. However, due to its relative novelty, little research has been done into how ATAC-seq data analysis should be conducted, especially in Drosophila. I developed data analysis pipelines for ATAC-seq in order to give a better understanding of what information this data gives us and its possible implications.
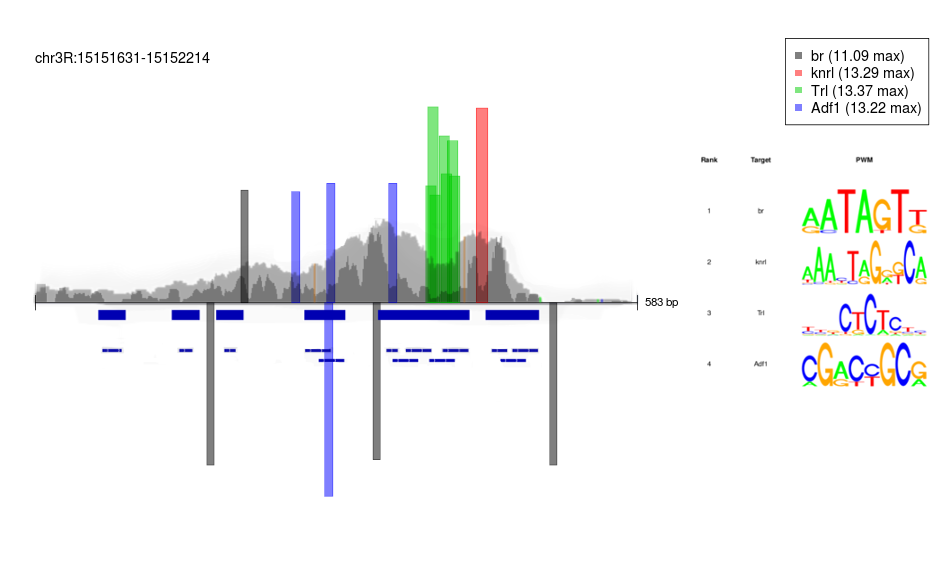
Transcription Factor Footprinting
During Drosophila development, cells from distinct imaginal discs (wing, leg, haltere) have seemingly identical open chromatin profiles at the same time point. This is surprising due to these cells resulting in very different structures. Thus, I was interested in ways of viewing chromatin accessibility at higher resolutions to see if these profiles are, in fact, identical. Specifically, I used de novo transcription factor footprinting with ATAC-seq data to find if subtle differences in DNA-binding protein interactions could reveal insight into how these cells differentiate.
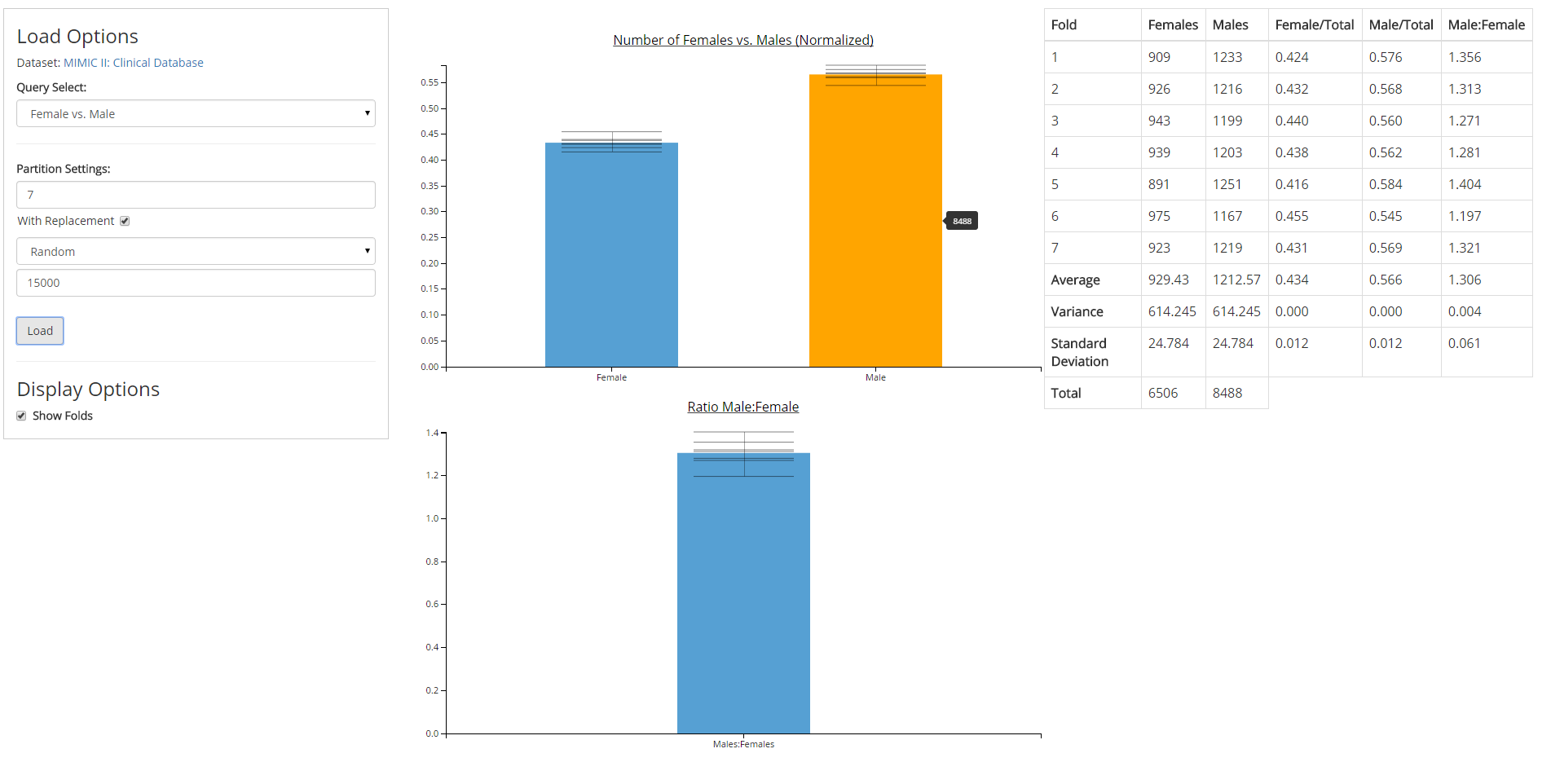
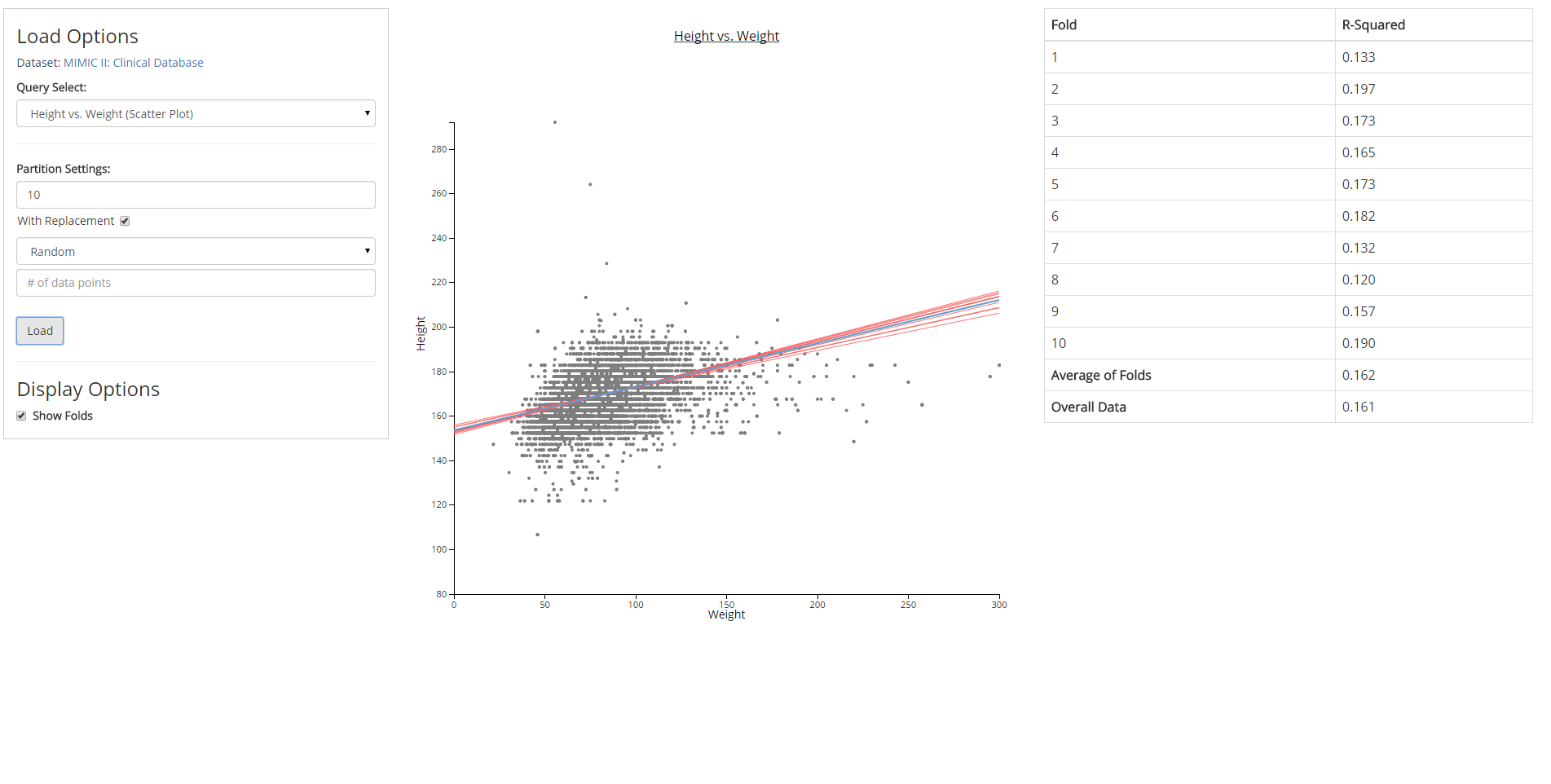
Directed by Dr. David Gotz, I built software prototypes for new data visualization and analysis methods developed by the Visual Analysis and Communication Laboratory (VACLab).
David Gotz, Brandon A. Price, Annie Chen. Visual Model validation via Inline Replication. Submitted to IEEE Visual Analytics Science and Technology (2015)